
Traditional Drug Development Process
How the process of drug development works and impacts overall health care benefit cost projections
May 2020Photo: iStock.com/LumiNola
Actuaries have often been considered the quantitative, objective sources of health care benefit cost projections. Lately, prescription drug benefit costs have been one of the fastest-growing components of health care benefit costs, thereby contributing to the overall growth of health care benefit costs in the United States (which the Society of Actuaries’ (SOA’s) 18|11 Initiative is seeking to address). One of the great complexities in projecting the prescription drug benefit costs for future benefit years is the ability to track new therapies in the pharmaceutical development pipeline and anticipate not only the timing of when they will come to market but also the market dynamics (including price) that will affect the utilization and cost-per-unit factors to drive their contribution to overall health care benefit costs. This article will help actuaries better understand the drug development process for traditional pharmaceuticals and how it impacts overall health care benefit cost projections.
We will begin by discussing the path of development and approval for traditional drugs. Traditional drugs and the newer specialty drugs are differentiated in several ways. However, since there are similarities in the development and approval process between the specialty and traditional drugs, the information in this article will apply to some specialty drugs. We will explore specialty drugs more deeply at a different time.
Drug Development Background
Since the beginning of humanity, we have tried to alleviate illnesses or symptoms of these illnesses. Up until recent times, we have turned toward nature to provide these alleviations, with mixed results. Traditional medicine still uses things from nature but in a much more scientific way than our ancestors did. In this article, we will discuss how we have transformed the process of identifying helpful natural compounds into scientifically recognized medicines. The process for scientific approval has changed through the years, as have the drugs themselves.
Drug ideas in the earlier days typically were initiated from active ingredients of traditional remedies or by serendipitous discoveries. In more recent times, drug discovery has included more definitive laboratorial progression steps: identifying and validating a target, generating assays to find lead compounds, and optimizing lead compounds to increase affinity and efficacy and to reduce potential side effects. As the process evolves to something more complex, the time and capital—which are required to develop a drug from an idea to laboratory bench work, to clinical trials and to a product—have also increased. Most sources for this research quoted 12 years or more as average development time.
At the same time, the types of researchers involved in the drug discovery and development process have expanded from chemists, physiologists and statisticians to include biochemists who study the chemistry of life processes, molecular biologists who study the molecules that make up living matter, toxicologists who investigate chemicals’ potential for harm and pharmacologists who look at how drugs work. As computing power becomes more accessible, computer scientists’ involvement also becomes more important in accessing, matching and analyzing data from, for example, chemical libraries or gene sequencing efforts.
Multiple funding mechanisms and partners are involved in the drug research and development process. Historically, the government and philanthropic organizations were the primary funders for early basic discovery research. Pharmaceutical companies and/or venture capitalists would take on the burden for funding drug discovery and late-stage development. Increased costs associated with inflation, navigating the regulatory pathway with novel products and technologies, and lawsuits have led to greater participation by venture capitalists. This is particularly the case for research on new technology platforms for target validation and associated therapeutics. A recent analysis of the 210 new molecular entities (NMEs) approved from 2010–2016 showed that National Institutes of Health (NIH) funding was directly or indirectly associated with all targets related to the 210 NMEs.1 Ultimately, the government and public funding (largely through NIH) continues to financially support basic research on nearly all drugs that are ultimately approved by the U.S. Food & Drug Administration (FDA).
One example of collaboration in drug discovery and development exists in the 2002 publishing of the human kinome, a subset of the human genome, through a collaboration between a biotechnology company and a research institute. These findings greatly impacted the treatment of cancer and other diseases. Another example pertains to patients with mutations within the JAK-3 kinase where research was done by NIH. Then, scientists at Pfizer, a large pharmaceutical manufacturer, spent several years discovering a compound that could treat the JAK-3 kinase mutation without causing severe side effects. Pfizer collaborated with the transplant center at Stanford University to test the drug with transplant recipients. Investigations were done for other treatment indications, and eventually the drug received FDA approval. These examples are described in “E, The Pathway from Idea to Regulatory Approval: Examples for Drug Development” in an Institute of Medicine report.
The more complex treatments that are being developed have increased the amount of spending needed for research and discovery. In 2016, the world spent more than $150 billion on research and development between government and business funding. The United States accounts for a large portion of that spending with estimates ranging from 58–65 percent of total spending.2,3
Research and Discovery
The early research and discovery process (see Figure 1) is characterized by a sequence of key events: therapeutic target identification and validation, identification of lead compounds and optimization of a lead compound to identify a development candidate.
Figure 1: Early Research and Discovery Process

Target Identification and Validation
A target is a macromolecular object (e.g., protein) that performs a biological function. It also has been referred to as a receptor. A drug target has a specific implication for a disease. Drugs are molecules or ligands that bind to receptors or drug targets. A good drug target should be safe and meet clinical and commercial requirements.
Drug target identification begins with identifying the function of possible therapeutic targets, either a protein or a gene that defines how proteins are structured (gene targets will lead to a specialty drug and will be discussed at a different time). The drug to be developed will be used to attack or modify the target protein that leads to the specific disease or condition needing a new treatment option. Most often, researchers find ideas for drug targets from academic research, scientific literature or bioinformatics data mining. The advantage of bioinformatics data mining is that it can also perform selection and prioritization in finding a potential disease target. As artificial intelligence (AI) methods emerge, the bioinformatics data mining processes may advance significantly.
After the targets are identified and selected, the molecular targets need to demonstrate the functional role of having therapeutic effect on the onset or progression of a disease. In a target validation process, experimental models and assays are developed to screen and evaluate the pharmacological link to the phenotype of interest. Validation using tool compounds (chemical genetics) or via genetic approaches can be conducted. These efforts will confirm not only the link to the disease of interest, but also that modulation of the target is safe.4,5
The target identification and validation process are iterative. In fact, pharmaceutical companies of all sizes are beginning to use AI in the drug discovery process either through their own internal programs or through collaboration with AI companies. Most experts expect AI tools to become more prevalent and more frequently used in the drug discovery process, thereby leading to quicker, cheaper and more effective drug discovery.6
A good example is the G Protein-Coupled Receptors (GPCRs), which constitute a large protein family of receptors from which approximately 34 percent of all FDA-approved drugs have come. Examples of drugs developed from GPCR protein families include citalopram, lisinopril and rivaroxaban.7
Lead Compound Screening
A lead compound is a new chemical with certain biological or pharmacological activity that has potential to be made into a therapeutic for the disease in question. To determine interactions with the target, researchers develop assays to screen presumptive series of compounds or small molecules, and experiment with multiple methods and technologies in those screenings. A smaller sample of molecules is then selected for its ability to actively engage with the target.
Two top experimental screening methods are high-throughput screening (HTS) and focused or knowledge-based screening. HTS uses automated robotics to quickly perform millions of assays to match the drug-like properties, with large libraries of compounds assuming no prior knowledge on the compounds. Focused screening involves narrowing compounds to a smaller subset that may have certain prior-known activity with the drug target through literature or patent precedents.8,9
Lead Compound Optimization
Researchers typically go through iterative rounds of synthesis and characterization to optimize the properties of lead compounds. Through these iterations, developers record detail measurement of how active and selective each core compound is to the target protein versus other nontarget proteins. They will select the one most actively binding to the target to be the lead compound.10,11
The optimization process tests not only the compounds potency and selectivity, but also the toxicity, safety, molecule mechanism and distribution. For any defects in a lead compound, such as low action intensity or specificity, inappropriate pharmacokinetic properties, strong toxic side effects, or chemical or metabolic instability, developers further modify the chemical structure to achieve a development candidate with balanced properties.
Development and Approval
The drug development and approval process includes five steps (see Figure 2).
Figure 2: Drug Development and Approval Process
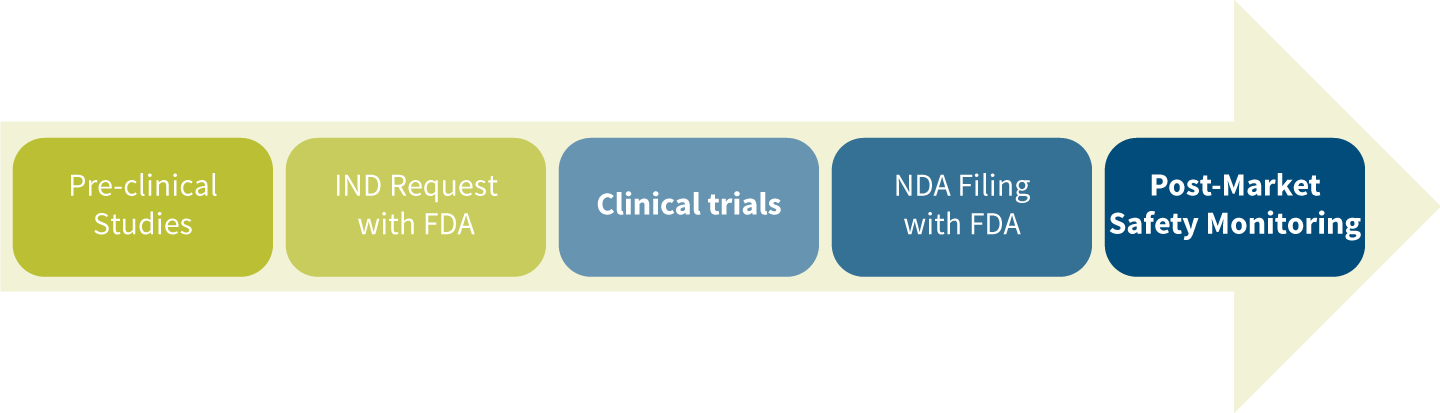
Pre-clinical Studies
Pre-clinical testing commonly divides into in vitro and in vivo testing. The time for preclinical testing often lasts many years. The purpose of pre-clinical trials is to obtain preliminary efficacy, toxicity, pharmacokinetic (what the body does to the drug) and pharmacodynamics (what the drug does to the body) information. Among other things, this information is used to determine the safe dose for human studies. In vitro testing examines the drug molecule interactions in test tubes and within the lab setting. In vivo testing involves testing the drug molecules on animal models and in other living cell cultures. The FDA will not let preclinical studies move into human trials without an establishment of safety. Examples of reasons drugs will not make it beyond pre-clinical studies include excessive toxicity such as carcinogenic (cancer-causing) or toxic effects on mammalian reproduction.12,13,14
Investigational New Drug (IND) Request With FDA
IND is a request for the FDA to authorize the start of human clinical trials. During this process, the FDA will scrutinize the results from the preclinical testing, look at side effects and other safety features of an experimental drug, examine the drugs’ chemical structure and how it is believed to work, and take a first look at the manufacturing process of the drug. Pharmaceutical companies must supply all pre-clinical study information and data for the FDA to consider approval of the IND.
- Drug developers need the application to ship an experimental drug across state lines before they obtain a drug marketing application approval.
- A patented drug’s 20-year exclusivity period begins upon IND’s approval and runs concurrently with clinical trials—thus the patent will always be less than 20 years by the time the drug gets FDA approval.
When a pharmaceutical company is pursuing a new indication for a drug that is already FDA-approved, an IND is not generally required, but instead, the company will submit a Supplemental New Drug Application (sNDA), which will include data for the drug on the new indication that is sought.
Clinical Trials
Phase 1 Human Clinical Studies
This stage of study involves looking at how a drug is absorbed and eliminated from the body, as well as what side effects it may cause and whether it’s producing the desired effect.
- Studies are normally conducted with a smaller group of a few dozen healthy volunteers.
- Doses with maximum performance that meet safety requirements are established at this step.
Phase 2 (Mid-stage) Clinical Studies
The main focus of this clinical studies step is safety. Short-term side effects of the drugs are closely monitored, with an increasing emphasis on whether a drug is working as expected and improving the condition.
- The patient pool widens from a few dozen patients to 100 or more patients.
- Patients are those who are afflicted by the disease in question.
- The most optimal dose, if multiple doses were tested, will most likely be established at this stage.
Phase 3 Clinical Studies
Tested drugs will be judged in phase 3 by following clearly defined guidelines from the FDA that define endpoints to determine if each study is a success or a failure. If drug developers anticipate success in this phase of studies, they will begin to think about how they’re going to ramp up production.
- Safety and efficacy play an important role in this stage.
- The patient pool involves a few hundred to thousands of patients.
- Phase 3 is the longest and costliest of all phases of the drug development process.
At any point in the clinical study phases, a drug can fail a clinical trial and be withdrawn from the process. A pharmaceutical company’s decision to end development can be based on a variety of reasons. Reasons for failure could be clinical and/or operational in nature. Some common reasons for failure include: lack of efficacy, issues with safety, poor study design or a lack of funding to complete a trial.
New Drug Application (NDA) Filing With FDA
Typically, when developers consider their phase 3 trial outcomes to be promising, they will file the NDA with the FDA. In situations such as a rare disease or a high unmet clinical need (particularly in the cancer disease states), an NDA may be filed prior to phase 3 trial data availability. For the NDA submission, developers need to include all research and safety data examined, and all animal and human clinical data collected. FDA reviewers look for these key factors in this process:
- Proposed labeling
- Safety updates
- Drug abuse information
- Patent information
- Any data from studies that may have been conducted outside the United States
- Institutional review board compliance information
- Directions for use
Under the standard approval process, if the NDA is accepted, the FDA can collect user fees and has the next 10 months to make a decision. The Prescription Drug User Fee Act (PDUFA) requires pharmaceutical companies to pay fees to fund the new drug approval process. The fees are available on the FDA website and shown in Figure 3.
Figure 3: FDA Fees
| User Fee Type | 2020 | 2019 |
| Application Fee – Clinical Data Required | $2,942,965 | $2,588,478 |
| Application Fee – No Clinical Data Required | $1,471,483 | $1,294,239 |
| Program Fee | $325,424 | $309,915 |
FDA has four opportunities that can expedite the review process. These processes were developed to speed the availability of drugs that treat serious conditions where currently there are no drug treatments or where new therapies may offer significant advantages over existing treatments. Each drug going through the review process may qualify for one or more of these designations:
- Fast Track designation
- Accelerated Approval
- Priority Review
- Breakthrough Therapy designation
Any of these designations are considered beneficial for a new drug application in that they highlight the potential to address an unmet clinical need and possibly get the drug on the market sooner. Both benefits may translate into higher revenue potential for the manufacturers.
The company developing the drug must request a Fast Track designation. The FDA will decide within 60 days based upon the determination if the new drug is filling an unmet medical need where no therapy currently exists or is potentially better than existing therapy. Fast Track designation benefits include more frequent meetings and communication with the FDA during the development and review process as well as eligibility for Accelerated Approval and Priority Review, if criteria are met.
The FDA instituted the Accelerated Approval regulations in 1992. These regulations allow surrogate or intermediate clinical endpoints to be used for efficacy measures for drugs in serious conditions with an unmet medical need. Using surrogate endpoints allows for less time for studies in conditions that may take many years to demonstrate real effects on how a patient survives, feels or functions.
Under the 1992 Prescription Drug User Fee Act (PDUFA), there is a two-tier system of review times: Standard Review and Priority Review. With a Priority Review designation, the FDA acts within six months versus the standard review time of 10 months. The FDA determines the review designation for each new drug application, although the applicant may also request Priority Review.
Breakthrough Therapy designation is requested by the drug company. The FDA may also suggest that the company consider submitting the request. The request is approved or declined within 60 days. Breakthrough Therapy designations are intended for drugs that treat serious conditions and where preliminary clinical evidence shows substantial improvement over available therapy based on one or more endpoints (such as surrogate endpoints, biomarkers and/or safety profile).15,16
Once a drug receives approval in the NDA process, it becomes immediately available for commercial production.
Post-Market Safety Monitoring
FDA’s monitoring of drug safety and other related issues through the Federal Food, Drug and Cosmetic Act (FD&C Act) continues after the drug is on the market. It conducts routine inspections of drug manufacturing facilities across the United States and overseas to ensure certain manufacturing practices are followed. Manufacturers or distributors normally will voluntarily recall defective products when they are discovered. There are three level-of-severity recalls from FDA. Class I is for products that cause the most serious health problems or death. Class II is for products that might cause a temporary health problem or a slightly less serious threat. Class III is for products that are unlikely to cause any adverse health reaction but violate FDA regulation.17
In addition to inspections, FDA also monitors drug advertisements and promotional labeling, and sets up MedWatch and Medical Product Safety Network (MedSun) for manufacturers, health professionals and consumers to report problems associated with approved drugs or medical devices. Any violation of the FD&C Act can result in a variety of actions specified by law. FDA actions can include any of the following: warning letters, seizure, injunction, criminal prosecution and criminal fines.
The goal of these post-market activities is to make sure the approved drugs continue to deliver effectively for their intended purposes and that consumers are informed as early as possible about any side effects or problems discovered and reported.
Drug Development Costs
According to a 2016 study by the Tufts Center for the Study of Drug Development,18 the estimated cost for pharmaceutical manufacturers to bring a new prescription medication from discovery to FDA approval per approved drug is $1.395 billion for out-of-pocket costs or $2.558 billion fully capitalized total cost (2013 U.S. dollars). There is some controversy on these figures. A more recent study19 found that the median cost of developing a single cancer drug is $648 million for out-of-pocket costs and $757 million in capitalized costs (2017 U.S. dollars). A 2019 study,20 estimated development costs of orphan (rare disease) and non-orphan drugs. They found that for each approved orphan drug, the out-of-pocket cost is $166 million and the capitalized cost is $291 million. For approved non-orphan drugs, the out-of-pocket costs per approved drug were $291 million and the capitalized costs per approved drug were $412 million. Part of the reason for the large variability in R&D costs is likely due to methodological differences. Given the large variation in R&D cost estimates, it is difficult to suggest that the price of a drug should be based upon the average R&D and production costs.
The reality is that in a capitalistic market, there are essentially no rules or laws defining how a drug company should price their drugs. When the FDA initially approves drugs, because it’s a “free market,” pharmaceutical companies typically select a price that they believe the market will bear. For drugs entering a competitive disease state (for example, diabetes or high cholesterol) with several comparable drugs already FDA-approved, generally the new drug will be priced similar to its competitors—unless, of course, the manufacturer thinks the new drug has a competitive and/or clinical advantage over the other drugs. In this case, it is likely the new drug will be launched at a premium price compared to its competitors. This is the dynamic situation that is common in the traditional drug space, which is perhaps better described as the old-fashioned pill-form medications. It even applies to injectable drugs such as insulin and the relatively newer injectable drugs such as Enbrel and Humira. These types of drugs developed with many therapeutic similarities to other existing drugs already on the market are often referred to as “me too” drugs.
Alternatively, we see drugs developed for rare diseases, in some cases, where there are no drug treatments currently available. Or there are examples where new drugs are developed with such dramatic improvement in clinical outcome (i.e., a cure for hepatitis C) that the new drug is considered a “first-in-class” treatment, which typically comes with a higher price.
Some may suggest that in the current market-driven system, prices are (or should be) set according to the value of the drug as measured by the improvement in disease, quality of life and/or other treatment cost avoidance. There may be some examples of this approach, but far more often, pricing continues to be driven by what the market is perceived to be able to bear, or by comparison to competitive treatment alternatives that are already on the market.
The Institute for Clinical and Economic Review (ICER) is a Boston-based institute that describes itself as “a trustworthy, independent source to help assess how valuable a new drug really is.” In 2015, ICER began its Emerging Therapy Assessment and Pricing Program aimed at developing value-based price benchmarks for newly approved drugs. Partially due to increased scrutiny by organizations such as ICER, there has been greater criticism of pricing when the cost-effectiveness of newer therapies exceeds industry-defined thresholds. Concerns have been raised regarding ICER’s definition of “value,” in that it is more short-term based and not as applicable to longer-term outcomes such as extending life or improving quality of life. Regardless, payers increasingly view ICER as a resource to be considered in making their formulary and budgetary decisions. Payers often push back on pharmaceutical manufacturers when they launch high-priced drugs that are not in line with the ICER cost-effectiveness evaluations.21
Actuaries should be familiar with ICER methodologies and findings in order to consider if and/or how those findings might integrate into actuarial health care benefit cost projections for different types of payers (health fully insured products, self-funded employer benefits, government program benefits, etc.). With greater understanding of ICER methodologies and findings, actuaries may be able to contribute to pharmaceutical manufacturer efforts to assess the viability of potential markets for the products they are researching and considering bringing to market far earlier in the development process—thereby improving investment and resource allocation efficiencies and helping bring to market the therapies that offer the most health and financial value to patients.
Acknowledgments: Many thanks to Dr. Natasja Brooijmans for her helpful firsthand insight and feedback into the real world of research and development.
References:
- 1. Cleary E. G., J. M. Beierlein, N.S. Khanuja, L.M. McNamee, and F. D. Ledley. 2018. Contribution of NIH Funding to New Drug Approvals. Proceedings of the National Academy of Sciences 115, no. 10:2,329–2,334. ↩
- 2. Organisation for Economic Co-operation and Development. 2019. Research and Development in the Pharmaceutical Sector. Health at a Glance 2019. ↩
- 3. The Association of the British Pharmaceutical Industry. Worldwide Pharmaceutical Company R&D Expenditure by Country. The Association of the British Pharmaceutical Industry (accessed April 17, 2020). ↩
- 4. Chen, J., X. Luo, H. Qiu, V. Macke, L. Sun, and X. Ouyang. 2018. Drug Discovery and Drug Marketing With the Critical Roles of Modern Administration. American Journal of Translational Research 10, no. 12:4,302–4,312. ↩
- 5. Hughes, J. P., S. Rees, S.B. Kalindjian, and K.L. Philpott. 2011. Principles of Early Drug Discovery. British Journal of Pharmacology 162, no 6:1,239–1,249. ↩
- 6. Fleming, N. 2018. How Artificial Intelligence is Changing Drug Discovery. Nature 557, no 7707:S55–S57. ↩
- 7. Institute of Medicine (U.S.) Committee on Conflict of Interest in Medical Research, Education and Practice; Bernard Lo, and Marylin J. Field, editors. 2009. Conflict of Interest in Medical Research, Education and Practice. Washington, D.C.: National Academies Press. ↩
- 8. Supra note 4. ↩
- 9. Supra note 5. ↩
- 10. Supra note 4. ↩
- 11. Supra note 5. ↩
- 12. Supra note 4. ↩
- 13. Supra note 5. ↩
- 14. Durrant, Jacob D. 2016. How Drugs Work. Durrant Lab. ↩
- 15. Supra note 4. ↩
- 16. Supra note 5. ↩
- 17. Kaehler, N., B. Adhikari, P.Y. Cheah, N. Day, D.H. Paris, M. Tanner, and C. Pell. 2019. The Promise, Problems and Pitfalls of Mass Drug Administration for Malaria Elimination: A Qualitative Study With Scientists and Policymakers. International Health 11, no. 3:166–176. ↩
- 18. DiMasi J.A., H.G. Grabowski, and R.W. Hansen. 2016. Innovation in the Pharmaceutical Industry: New Estimates of R&D Costs. Journal of Health Economics 47:20–33. ↩
- 19. Prasad, V., and S. Mailankody. 2017. Research and Development Spending to Bring a Single Cancer Drug to Market and Revenues After Approval. JAMA Internal Medicine 177, no. 11:1,569–1,575. ↩
- 20. Jayasundara, K., A. Hollis, M. Krahn, et al. 2019. Estimating the Clinical Cost of Drug Development for Orphan Versus Non-orphan Drugs. Orphanet Journal of Rare Diseases 14, no. 12. ↩
- 21. Cohen, J. 2019. ICER’s Growing Impact on Drug Pricing and Reimbursement. Forbes. ↩
Copyright © 2020 by the Society of Actuaries, Chicago, Illinois.